Signed in as:
filler@godaddy.com
The R6/2 Transgenic Mouse Model is one of the most widely used and well-characterized models for studying Huntington’s disease (HD). This model carries a truncated form of the mutant huntingtin (HTT) gene with an expanded CAG repeat, which is responsible for the neurodegenerative processes seen in Huntington’s disease. The R6/2 mouse exhibits many of the hallmark features of HD, including progressive motor dysfunction, cognitive decline, and neurodegeneration.
Key Features of the R6/2 Model:
This model continues to be instrumental in advancing our understanding of Huntington's disease and developing new treatments. Naason Science has completed validation of R6/2 mouse line for the study of HD; the model paradigm has a complete functional and behavioral battery, imaging (MRI/MRS) and histological and molecular analysis of various regions of brain.
Naason Science model assays:
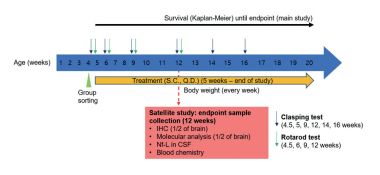
Study design

The TG (zQ175) Mouse Model of Huntington’s Disease is a widely utilized transgenic model that carries the full-length human mutant huntingtin (HTT) gene with an expanded CAG repeat, closely mimicking the genetic mutation found in HD patients. The zQ175 model represents a slower progression of the disease compared to other models, making it an excellent tool for studying long-term therapeutic interventions and disease mechanisms.
Key Features of the zQ175 Model:
This model is crucial for advancing our understanding of the molecular mechanisms of Huntington’s disease and for evaluating promising therapeutic interventions in a setting that closely mirrors human HD pathology.